
脊髄性筋萎縮症(指定難病3)
1. 「脊髄性筋萎縮症」とはどのような病気ですか
脊髄性筋萎縮症(spinal muscular atrophy: SMA)とは、脊髄の運動ニューロン(脊髄前角細胞)の病変によって起こる 神経原性の筋萎縮症 で、筋萎縮性側索硬化症(ALS)と同じ運動ニューロン病の範疇に入る病気です。体幹や四肢の筋力低下、筋萎縮を進行性に示します。小児期に発症するI型:重症型(別名:ウェルドニッヒ・ホフマンWerdnig-Hoffmann病)、II型:中間型(別名:デュボビッツDubowitz病)、III型:軽症型(別名:クーゲルベルグ・ウェランダーKugelberg-Welander病)と、成人期に発症するIV型に分類されます(表1)。主に小児期に発症するSMAは第5染色体に病因遺伝子を持つ常染色体潜性遺伝(劣性遺伝)性疾患ですが、成人発症のSMA IV型は遺伝子的に複数の成因の混在が考えられます。

2. この病気の患者さんはどのくらいいるのですか
SMAの有病率は10万人あたり1〜2人です。発生率は、出生2万人に対して1人前後と言われています。I型の発生率は4万人に1人です。成人発症のIV型はALSより少ないです。
3. この病気はどのような人に多いのですか
男女差はありません。I型は乳児期、II型は乳児期から幼児期、III型は幼児期から小児期、IV型は成人期に発症します。
4. この病気の原因はわかっているのですか
SMAの原因遺伝子は運動神経細胞生存(survival motor neuron:略してSMN)遺伝子です。第5染色体(5q13という部位)に存在しており、神経細胞 アポトーシス 抑制蛋白(neuronal apoptosis inhibitory protein:略してNAIP)遺伝子は 修飾遺伝子 です。SMN遺伝子の近傍には、SMN遺伝子とは16か所の20塩基対が異なっている遺伝子が存在しています。SMN遺伝子のことはSMN1遺伝子、近傍の遺伝子はSMN2遺伝子と名づけられています。SMN1遺伝子にてSMN蛋白質が作られますがSMN2遺伝子からは僅かのSMN蛋白質しか作られません。SMAにおいてはSMN1遺伝子の変異により、SMN1遺伝子由来のSMN蛋白質は作られず、この僅かのSMN2遺伝子由来のSMN蛋白質しかない状態となります。SMN2遺伝子の数はSMN2遺伝子のコピー数で表しますが、父親から1コピー、母親から1コピーを受けた2コピーより3コピーの方が、3コピーより4コピーの方が軽症となる傾向があります。SMN蛋白質は多くの組織、細胞で広く発現して、様々な細胞プロセスに関与しています。NAIPは昆虫細胞のアポトーシスを抑制する蛋白質と構造が似ているため、神経細胞のアポトーシスを抑制する蛋白質と考えられています。I、II型の95%にSMN遺伝子欠失が認められ、III型の約半数、IV型の1-2割においてSMN遺伝子 変異 が認められます。
5. この病気は遺伝するのですか
SMN1遺伝子変異を示すSMAは 常染色体潜性遺伝(劣性遺伝) 形式を示します。すなわち、父親由来のSMN1遺伝子と母親由来のSMN1遺伝子が共に変異を示している場合に、その子はSMAになります。父親由来または母親由来の遺伝子がどちらか1つだけ変異している場合は全く無症状であり、この場合を 保因者 といいます。保因者は生涯、症状がありません。保因者同士の結婚の場合、お子さんがSMAになる可能性は1/4(25%)です。I型の保因者の頻度は欧米では60〜80人に1人、II型、III型は76〜111人に1人といわれていますが、日本では欧米より少ないようです。保因者の頻度を100人に1人と仮定すると、保因者同士の結婚は1/100×1/100=1/10,000であり、お子さんがSMAとなる可能性は1/10,000×1/4=1/40,000となります。患者さん本人のお子さんがSMAになる可能性は1×1/100×1/2=1/200、患者さんの症状のない兄弟姉妹のお子さんがSMAになる可能性は2/3×1/100×1/4=1/600となり、遺伝病の発生頻度(1-2%)や先天異常症の発生頻度(数%)より低いです。
遺伝に関わる相談は、全国の大学病院に遺伝子医療部門があり、臨床遺伝専門医の 遺伝カウンセリング を受け、遺伝子検査も受けることができます。
6. この病気ではどのような症状がおきますか
全ての型で筋力低下と筋萎縮を示し、深部 腱反射 の減弱・消失が見られます。
I型は生後6カ月ごろまでに発症、運動発達が停止し、体幹を動かすこともできません。支えなしに座ることができず、哺乳困難、 嚥下困難 、誤嚥、呼吸不全を伴います。舌の細かい振え(線維束性収縮)がみられます。後述のような治療薬のなかった時代には、人工呼吸器を用いない場合、死亡年齢は平均6〜9カ月、95%は18カ月までに死亡するといわれておりました。生命を救うためには、多くの例で気管内挿管や気管切開と人工呼吸管理が必要でありました。
II型は支えなしに立ったり、歩いたりすることができません。舌の 線維束性収縮 、手指の振戦がみられます。成長とともに関節拘縮と側弯が著明になります。また、上気道感染に引き続いて、肺炎や 無気肺 になり、呼吸不全に陥ることがあります。
III型では立ったり歩いたりできていたのに、転びやすい、歩けない、立てないという症状がでてきます。次第に、上肢の挙上も困難になります。発症は幼児期、小児期です。歩行不可能になる時期は幼児期から成人期にわたり、生涯歩行している方もいます。小児期以前の発症ではII型と同様に側弯が生じます。
IV型は成人発症です。側弯は見られませんが、発症年齢が遅いほど進行のスピードは緩やかです。下位運動ニューロンのみの障害であり、ALSが上位ニューロンも障害されるのと比較されます。
7. この病気にはどのような治療法がありますか
本症では世界的に治療薬の開発と治験(臨床試験)が実施され、わが国でも以下の3つの疾患修飾治療薬が承認されました(表2)。
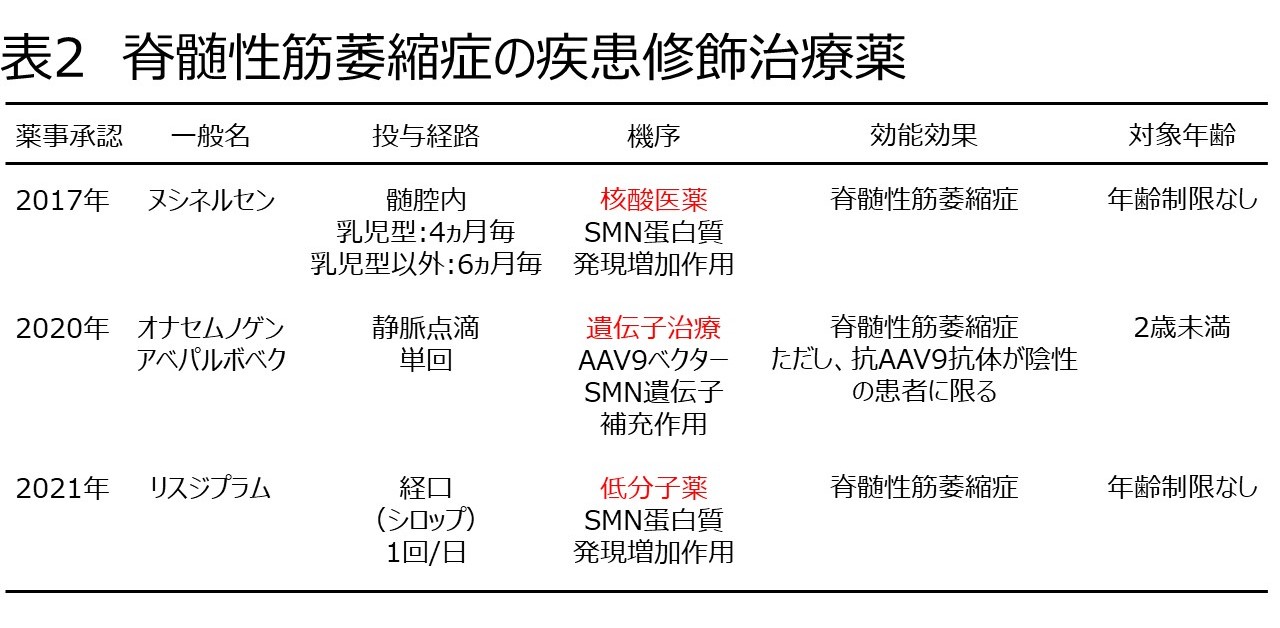
ヌシネルセンはSMN蛋白質を産生する機序をもつ核酸医薬品です。腰椎穿刺の要領で髄腔内投与をします。負荷投与後は乳児型では4カ月間隔、乳児型以外では6カ月間隔の投与です。オナセムノゲンアベパルボベクはAAV9(アデノ随伴ウイルス血清型9)ベクターにSMN遺伝子を組込んだ遺伝子治療の薬剤で2歳未満が対象です。1回1時間かけて点滴する静脈注射です。体内にAAV9に対する抗体ができるため繰返し投与はできません。リスジプラムもSMN蛋白質を産生する機序をもつ低分子薬です。経口投与薬(イチゴ味シロップ)で、毎日1回内服します。
新しい治療薬による治療を受ける方が増えている一方で、従来の「ケア」の概念も重要です。乳児期に発症するI、II型では、哺乳や 嚥下 が困難な場合、経管栄養や胃瘻が必要です。また、呼吸器感染、無気肺を繰り返す事は 予後 を大きく左右します。鼻マスク人工換気法(NPPV)は有効と考えられますが、乳児への使用には多くの困難を伴います。呼吸器感染時には、カフアシストの使用や、肺の理学療法による排痰ドレナージが有効です。また、筋力にあわせた運動訓練、関節拘縮の予防などのリハビリテーションが必要です。III型では歩行可能な状態をなるべく長期に維持できるように、また関節拘縮の予防のためにも、リハビリテーションを行い、装具の使用などを検討します。小児神経医、脳神経内科医、整形外科医、理学療法士の連携が必要です。
HAL医療用下肢タイプは本疾患における治験結果(https://doi.org/10.1186/s13023-021-01928-9)に基づき、2015年医療機器承認され、2016年から歩行運動処置(ロボットスーツによるもの)として保険適用されています。特定機能病院や多くの急性期病院等のDPC対象病院では、外来通院でのみ保険算定が認められていましたが、2022年から入院診療においてもDPCの包括外での算定が可能となりました。
8. 新生児スクリーニング検査で「陽性」となった場合には、どうしたらよいですか
新生児スクリーニング検査は確定検査ではありませんので、すぐに専門医を受診して、確定検査として遺伝子検査を受けてください。遺伝子検査の結果、SMAと診断された場合には、発症前(SMAの症状が出る前)にヌシネルセンまたはオナセムノゲンアベパルボベク、リスジプラムの治療を受けることで発病を抑えたり、軽症化させることが可能です。発症前に治療を受けた場合の有効性がこれらの薬剤の治験で報告されています。もし、既に症状が出ていても、治療は早ければ早いほど有効です。お子さんが遺伝子検査の結果、I型やⅡ型になる可能性がある、つまり、SMN1遺伝子が0コピーで、かつSMN2遺伝子が2コピーまたは3コピーという報告を受けた場合には、SMAの症状がなくても、大至急の治療開始が推奨されています。また、症状が出現していなくても、遺伝子検査でSMAの診断を受けた場合には、小児慢性特定疾病または指定難病の申請をすると、医療費助成を受けることができます。一方、5-10%の頻度で、新生児スクリーニングで「陰性」であってもSMAを発症する場合があります。SMN1遺伝子が1コピーのため、もうひとつのSMN1遺伝子変異があっても、新生児スクリーニングでは「陰性」となってしまうためです。
9. この病気はどういう経過をたどるのですか
疾患修飾治療薬により、病気の経過は変わりつつあります。自然経過では症状は徐々に進行する場合が多いですが、症状が一時期、進行した後、停止するような場合もあります。I、II型では、成長と共に運動機能の獲得の時期もあります。呼吸器感染症にて肺炎、無気肺となると急速に進行したり、人工呼吸管理が必要となる場合があります。
10. この病気は日常生活でどのような注意が必要ですか
I、II型の乳幼児では、咳の力が弱い、呼吸器感染を繰り返す、哺乳や食事摂取で疲労を示す、体重が増えないなどの医療的管理が必要な徴候に注意を払う必要があります。適切な時期の予防接種が必要です。24カ月齢以下の重症化リスクが高い神経筋疾患としてRSウイルスワクチンが保険適応となりました。一方で、食欲の良好なII、III型の幼児では、肥満にならない注意も必要です。側弯や関節拘縮を防ぐために、座位保持装置や良好な姿勢の管理が必要です。理学療法士に相談をすると良いでしょう。自力での移動が困難な方の生活において車椅子が必要です。上肢筋力も弱いため、電動車椅子を必要とします。II型のお子さんは、3歳になると電動車椅子を動かすことができ、家や外で家族や友達と一緒に活動できるようになります。
発症前に治療を受けて、発症が抑えられている方においても、疾患修飾治療薬の長期の有効性や安全性に関しての経験が世界的にも十分ではないため、定期的な専門医の診察が推奨されます。
11. 次の病名はこの病気の別名又はこの病気に含まれる、あるいは深く関連する病名です。 ただし、これらの病気(病名)であっても医療費助成の対象とならないこともありますので、主治医に相談してください。
ウェルドニッヒ・ホフマン病 Werdnig-Hoffmann病
デュボヴィッツ病 Dubowitz病
クーゲルベルグ・ウェランダー病 Kugelberg-Welander病
12.病気に関する資料・関連リンク
脊髄性筋萎縮症診療マニュアル. 脊髄性筋萎縮症診療マニュアル編集委員会. 2012. pp150. 金芳堂. 京都
脊髄性筋萎縮症(SMA)診療の手引き. 脊髄性筋萎縮症(SMA)診療の手引き編集委員会.2022.pp280. メディカルレビュー社. 東京
脊髄性筋萎縮症に対する新生児マススクリーニングの手引き. 日本小児神経学会編集.2023. pp29. https://www.childneuro.jp/
SMARTコンソーシアム(脊髄性筋萎縮症の治療を目指す患者登録システム). http://www.sma-rt.org/
SMA(脊髄性筋萎縮症)家族の会. http://www.sma-kazoku.net/