
両大血管右室起始症(指定難病216)
1.両大血管右室起始症とは?
正常の心臓では、左室からは大動脈が起始し、右室からは肺動脈が起始しますが、両大血管右室起始症では、大動脈と肺動脈の両方の大血管の一つともう一つの半分(50%ルール)以上が右室から起始している病気です。心室中隔に大きな穴(心室中隔欠損)を伴い、大血管の位置関係や心室中隔欠損孔の位置関係により、大きくは、大動脈弁下型心室中隔欠損(図1左、正常大血管型)、肺動脈弁下型心室中隔欠損(図1右、大血管転位型)に分類されています。そのほか、稀なタイプとして両大血管下型心室中隔欠損と遠隔型心室中隔欠損があります。
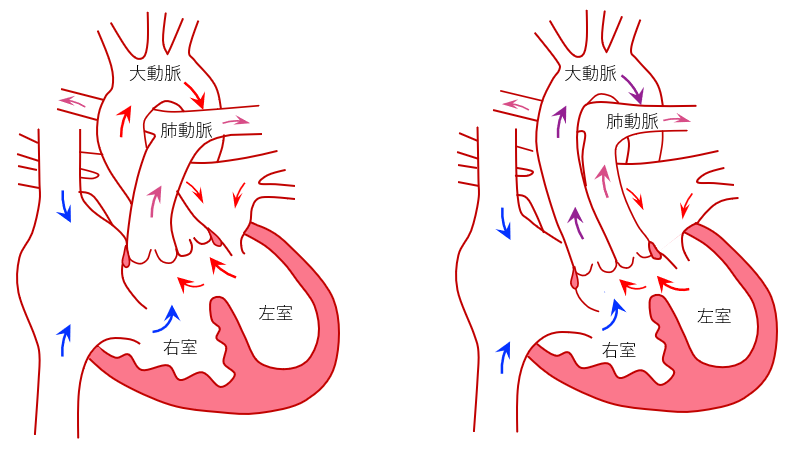
図1:両大血管右室起始の分類、左;大動脈弁下型心室中隔欠損、右;肺動脈弁下型心室中隔欠損
2. この病気の患者さんはどのくらいいるのですか。
先天性 心疾患の約1%(出生約10,000人に1人)とされています。
3. この病気はどのような人に多いのですか。
両大血管右室起始症が多い家系や遺伝性症候群はあまりありませんが、18トリソミーの患者さんには比較的頻度が高く認められます。
4. この病気の原因はわかっているのですか。
胎児期のできはじめの心臓の出口(右心室の流出路)には一つの太い血管(総動脈幹)が繋がっています。母体内で心臓の発生が進むと、右室流出路が心臓の左側に大きく移動するとともに、総動脈幹では内部にらせん状の仕切りができてきて大動脈と肺動脈が分かれるようになります。この結果、左側の後にできる大動脈が新たに左心室と交通するようになります。この過程、すなわち、右室流出路の左側への移動もしくは総動脈幹のらせん分割に異常があると、両大血管右室始起が発症します。
5. この病気は遺伝するのですか。
両大血管右室起始症では、明らかに強い遺伝性は認められていません。他の疾患を含めた先天性心疾患のきょうだいでの発症率は通常の1%より少し高くなるとされています。一般に先天性心疾患の親から子へ何らかの先天性心疾患が遺伝する確率は、父親で1-3%程度、母親で2-12%程度とされています。
6. この病気ではどのような症状がおきますか。
大動脈下型心室中隔欠損(正常大血管型)で肺動脈 狭窄 を伴わないお子さんでは、左心室から右心室へ大量の血液が短絡しますので、新生児や乳児では、多呼吸、陥没呼吸、哺乳不良、体重増加不良、発汗などの心不全症状が認められます。肺動脈狭窄を伴う場合はファロー四徴のような チアノーゼ が新生児より見られ、その程度は肺動脈狭窄の程度により異なります。
肺動脈弁下型心室中隔欠損(大血管転位型)では、基本的に新生児期よりチアノーゼが見られます。ただし肺動脈狭窄を伴わない場合には、心不全症状が目立つこともあります。肺動脈狭窄を伴う場合は、新生児期より著しいチアノーゼが見られます。
7. この病気にはどのような治療法がありますか?
大動脈弁下型心室中隔欠損(正常大血管型)で肺動脈狭窄を伴わないお子さんでは、左心室の血液が大動脈にスムーズに流れるようにする形で心室中隔欠損孔を閉鎖します。肺動脈狭窄を伴う場合は、肺動脈狭窄の程度が強ければ、乳児期早期にブラロック・トーシッヒ短絡手術を行い、肺血流を増やしてチアノーゼを改善し、成長を待ってから乳児期後期に心内修復術(心室中隔欠損孔閉鎖と右室流出路狭窄解除術)を行います。
肺動脈弁下型心室中隔欠損(大血管転位型)で肺動脈狭窄をともなわない場合は、新生児期から乳児期早期に動脈スイッチ手術(ジャテネ手術)を行います。強い肺動脈狭窄をともなう場合は、新生児期にブラロック・トーシッヒ短絡手術を行い肺血流を増やし、幼児期早期にラステリー手術を行って、心室中隔欠損孔を閉鎖するとともに、人工血管により右心室から肺動脈へ血液を導きます。
遠隔型心室中隔欠損の一部の患者さんで、大動脈が左心室から遠く離れている場合には2心室修復手術ができず、1心室修復術であるフォンタン型手術を行う場合があります。
8. この病気はどういう経過をたどるのですか。
肺動脈狭窄を伴わない大動脈弁下型心室中隔欠損(正常大血管型)のお子さんでは、一般に単純な心室中隔外科手術後のように 予後 は良好で生活制限も必要でないことが多いです。肺動脈狭窄を伴うお子さんでは、ファロー四徴の術後に準じます。 生命予後 は比較的良好ですが、問題となる程度の肺動脈弁閉鎖不全や狭窄が遺残したお子さんでは、思春期や成人期に再手術が必要になることがあります。
肺動脈狭窄を伴わない肺動脈弁下型心室中隔欠損(大血管転位型)のお子さんでは、完全大血管転位のジャテネ手術後に準じます。近年、生命予後は比較的良好になりましたが、術後の肺動脈狭窄、大動脈弁閉鎖不全、冠動脈障害などの 重篤 な続発症が起こり得ますので、術後も慎重な経過観察が必要です。再手術や カテーテル治療 の可能性があります。
肺動脈狭窄を伴うお子さんでは、ラステリー手術後の続発症である導管狭窄、導管閉鎖不全が高率に認められます。再手術やカテーテル治療の必要性があります。
遠隔型心室中隔欠損などでフォンタン型手術を行った場合は、単心室のフォンタン型手術後の経過に準じます。
9. この病気は日常生活でどのような注意が必要ですか。
術後の遺残症および続発症の状態と程度によります。心不全や問題となるような不整脈のないお子さんでは、激しい運動は少し注意が必要ですが、概ね学校での体育活動は可能です。無理をしない範囲でのクラブ活動も可能なことが多いです。動脈スイッチ手術やラステリー手術などの複雑な手術を行ったお子さんでは、遺残および続発する肺動脈弁狭窄や閉鎖不全、大動脈弁閉鎖不全の程度、不整脈の性質と頻度により、学校での運動制限や生活制限が必要になってきます。主治医の指示に従って定期的な検査を受け、必要であれば薬を服用して、規則正しい無理のない生活をするようにしましょう。
10. 次の病名はこの病気の別名又はこの病気に含まれる、あるいは深く関連する病名です。 ただし、これらの病気(病名)であっても医療費助成の対象とならないこともありますので、主治医に相談してください。
該当する病名はありません。
11. この病気に関する資料・関連リンク
① 小児・成育循環器学(改訂第2版). 日本小児循環器学会編集. 診断と治療社, 2024.
② 日本成人先天性心疾患学会ホームページ総合・連携認定施設一覧
https://www.jsachd.org/specialist/list-facility/
③ 成人先天性心疾患診療ガイドライン(2017年改訂版)
https://www.j-circ.or.jp/cms/wp-content/uploads/2017/08/JCS2017_ichida_h.pdf
④ 先天性心疾患並びに小児期心疾患の診断検査と薬物療法ガイドライン(2018年改訂版)
https://www.j-circ.or.jp/cms/wp-content/uploads/2020/02/JCS2018_Yasukochi.pdf
⑤ 心疾患患者の妊娠・出産の適応、管理に関するガイドライン(2018年改訂版).
https://www.j-circ.or.jp/cms/wp-content/uploads/2018/06/JCS2018_akagi_ikeda.pdf
⑥ 先天性心疾患術後遠隔期の管理・侵襲的治療に関するガイドライン(2022年改訂版)
https://www.j-circ.or.jp/cms/wp-content/uploads/2022/03/JCS2022_Ohuchi_Kawada.pdf
| 研究班名 | 先天性心疾患を主体とする小児期発症の心血管難治性疾患の救命率の向上、円滑な移行医療、成人期以降の予後改善を目指した総合的研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和6年11月(名簿更新:令和6年7月) |